Baseline Model - ECFPs and Random Forest¶
Introduction¶
There are many ways to represent molecules for machine learning.
We will go through one of the simplest: ECFPs [1] and Random Forest. This technique is surprisingly powerful, and on previous benchmarks often gets uncomfortably close to the state of the art.
First molecule graphs are broken into bags of subgraphs of varying sizes.
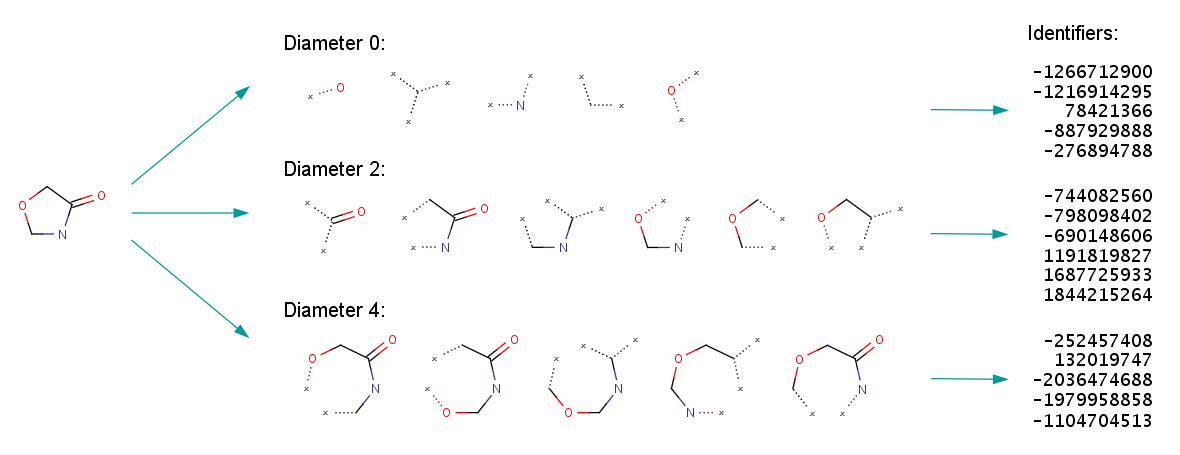
Then the bag of subgraphs is hashed into a bit vector
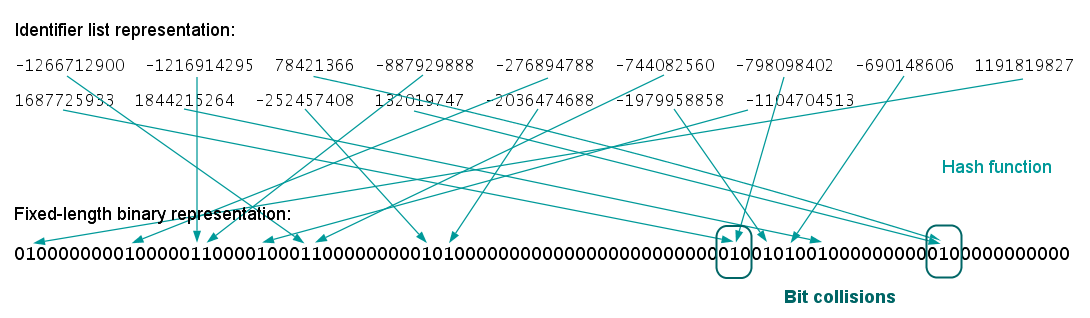
This can be thought of as analogous to the hashing trick [2] on bag of words for NLP problems, from the days before transformers.
RDKit, an open-source cheminformatics tool, is used for generating ECFP features. It facilitates the creation of hashed bit vectors, streamlining the process. We can install it as follows:
The training set is pretty big, but we can treat the parquet files as databases using duckdb. We will use this to sample down to a smaller dataset for demonstration purposes. Lets install duckdb as well.
Data Preparation¶
The training and testing data paths are defined for the .parquet files. We use duckdb to scan search through the large training sets. Just to get started lets sample out an equal number of positive and negatives.
This query selects an equal number of samples where binds equals 0 (non-binding) and 1 (binding), limited to 30,000 each, to avoid model bias towards a particular class.
import duckdb
import pandas as pd
train_path = '/dfs6/pub/ddlin/projects/belka_del/data/raw/train.parquet'
test_path = '/dfs6/pub/ddlin/projects/belka_del/data/raw/test.parquet'
con = duckdb.connect()
df = con.query(f"""(SELECT *
FROM parquet_scan('{train_path}')
WHERE binds = 0
ORDER BY random()
LIMIT 30000)
UNION ALL
(SELECT *
FROM parquet_scan('{train_path}')
WHERE binds = 1
ORDER BY random()
LIMIT 30000)""").df()
con.close()
FloatProgress(value=0.0, layout=Layout(width='auto'), style=ProgressStyle(bar_color='black'))
df.head()
| id | buildingblock1_smiles | buildingblock2_smiles | buildingblock3_smiles | molecule_smiles | protein_name | binds | |
|---|---|---|---|---|---|---|---|
| 0 | 60621538 | Cc1cccc(NC(=O)OCC2c3ccccc3-c3ccccc32)c1C(=O)O | N#Cc1c(N)cccc1F | Nc1ccc2cc(Br)ccc2c1 | Cc1cccc(Nc2nc(Nc3ccc4cc(Br)ccc4c3)nc(Nc3cccc(F... | HSA | 0 |
| 1 | 227061691 | O=C(Nc1nc2ccc(C(=O)O)cc2s1)OCC1c2ccccc2-c2ccccc21 | Cc1sc(CCN)nc1-c1ccccc1.Cl.Cl | Cc1cc(=O)oc2cc(N)ccc12 | Cc1sc(CCNc2nc(Nc3ccc4c(C)cc(=O)oc4c3)nc(Nc3nc4... | HSA | 0 |
| 2 | 41105038 | COc1nccc(C(=O)O)c1NC(=O)OCC1c2ccccc2-c2ccccc21 | NCc1c(F)cccc1N1CCCC1 | CS(=O)(=O)Nc1ccc(-c2csc(N)n2)cc1 | COc1nccc(C(=O)N[Dy])c1Nc1nc(NCc2c(F)cccc2N2CCC... | HSA | 0 |
| 3 | 7263624 | C=CCC(NC(=O)OCC1c2ccccc2-c2ccccc21)C(=O)O | NCC(O)COc1cccc(Cl)c1Cl | CCOC(=O)c1c[nH]nc1N | C=CCC(Nc1nc(NCC(O)COc2cccc(Cl)c2Cl)nc(Nc2n[nH]... | BRD4 | 0 |
| 4 | 261575461 | O=C(O)C[C@@H]1CCCN1C(=O)OCC1c2ccccc2-c2ccccc21 | CCOC(=O)c1cnc(N)cn1 | Cn1c(=O)cc(N)[nH]c1=O | CCOC(=O)c1cnc(Nc2nc(Nc3cc(=O)n(C)c(=O)[nH]3)nc... | HSA | 0 |
Feature Preprocessing¶
Lets grab the smiles for the fully assembled molecule molecule_smiles and generate ecfps for it. We could choose different radiuses or bits, but 2 and 1024 is pretty standard.
import rdkit
from rdkit import Chem
from rdkit.Chem.rdFingerprintGenerator import GetMorganGenerator
print(f"RDKit Version: {rdkit.__version__}")
mol = Chem.MolFromSmiles("CCO")
fpgen = GetMorganGenerator(radius=2, fpSize=1024)
print(f"\nGenerator object type: {type(fpgen)}")
print("\n--- Available Methods on fpgen object ---")
# This will print a list of all valid methods. Look for something similar to 'GetFingerprint...'.
print([method for method in dir(fpgen) if not method.startswith('_')])
RDKit Version: 2025.03.3 Generator object type: <class 'rdkit.Chem.rdFingerprintGenerator.FingerprintGenerator64'> --- Available Methods on fpgen object --- ['GetCountFingerprint', 'GetCountFingerprintAsNumPy', 'GetCountFingerprints', 'GetFingerprint', 'GetFingerprintAsNumPy', 'GetFingerprints', 'GetInfoString', 'GetOptions', 'GetSparseCountFingerprint', 'GetSparseCountFingerprints', 'GetSparseFingerprint', 'GetSparseFingerprints']
from rdkit import Chem
# Import the modern fingerprinting API
from rdkit.Chem.rdFingerprintGenerator import GetMorganGenerator
from sklearn.ensemble import RandomForestClassifier
from sklearn.model_selection import train_test_split
from sklearn.metrics import average_precision_score
from sklearn.preprocessing import OneHotEncoder
# Convert SMILES to RDKit molecules
df['molecule'] = df['molecule_smiles'].apply(Chem.MolFromSmiles)
# Generate ECFPs using the new MorganGenerator API and the CORRECT method name
def generate_ecfp(molecule, radius=2, bits=1024):
if molecule is None:
return None
# Create a generator object for Morgan fingerprints
fpgen = GetMorganGenerator(radius=radius, fpSize=bits)
# Use the correct method, .GetFingerprint(), as revealed by dir(fpgen)
return list(fpgen.GetFingerprint(molecule))
# This line will now execute successfully.
df['ecfp'] = df['molecule'].apply(generate_ecfp)
print("ECFP generation complete.")
ECFP generation complete.
Train Model¶
# One-hot encode the protein_name
onehot_encoder = OneHotEncoder(sparse_output=False)
protein_onehot = onehot_encoder.fit_transform(df['protein_name'].values.reshape(-1, 1))
# Combine ECFPs and one-hot encoded protein_name
X = [ecfp + protein for ecfp, protein in zip(df['ecfp'].tolist(), protein_onehot.tolist())]
y = df['binds'].tolist()
# Split the data into train and test sets
X_train, X_test, y_train, y_test = train_test_split(X, y, test_size=0.2, random_state=42)
# Create and train the random forest model, using all available cores (n_jobs=-1)
rf_model = RandomForestClassifier(n_estimators=100, random_state=42, n_jobs=-1)
rf_model.fit(X_train, y_train)
# Make predictions on the test set
y_pred_proba = rf_model.predict_proba(X_test)[:, 1] # Probability of the positive class
# Calculate the mean average precision
map_score = average_precision_score(y_test, y_pred_proba)
print(f"Mean Average Precision (mAP): {map_score:.2f}")
Mean Average Precision (mAP): 0.96
Look at that Average Precision score. We did amazing!
Actually no, we just overfit. This is likely recurring theme for this dataset. It is easy to predict molecules that come from the same corner of chemical space, but generalizing to new molecules is extremely difficult.
Test Prediction¶
The trained Random Forest model is then used to predict the binding probabilities. These predictions are saved to a CSV file, which serves as the submission file for the competition.
import os
import pandas as pd
from multiprocessing import Pool, cpu_count
import gc
from tqdm import tqdm
from rdkit import Chem
from rdkit.Chem.rdFingerprintGenerator import GetMorganGenerator
from rdkit.Chem import AllChem # Make sure to import AllChem
# --- Configuration ---
ECFP_RADIUS = 2
ECFP_NBITS = 1024 # Matching the bits used in training
def smiles_to_ecfp_worker(smiles):
"""A dedicated worker function that uses the older AllChem API."""
try:
mol = Chem.MolFromSmiles(smiles)
if mol:
# Reverting to the AllChem function compatible with older RDKit
return list(AllChem.GetMorganFingerprintAsBitVect(mol, ECFP_RADIUS, nBits=ECFP_NBITS))
return [0] * ECFP_NBITS
except:
return [0] * ECFP_NBITS
# --- Main Processing Script ---
test_file = '/dfs6/pub/ddlin/projects/belka_del/data/raw/test.csv'
output_file = 'ecfps_submission.csv'
num_processes = cpu_count() - 2 if cpu_count() > 2 else 1
print(f"Using {num_processes} worker processes.")
if os.path.exists(output_file):
os.remove(output_file)
chunk_size = 100000
# --- TQDM SETUP: Get total number of rows for the main progress bar ---
print("Calculating total number of molecules...")
total_rows = sum(1 for row in open(test_file, 'r')) - 1 # Subtract 1 for header
print(f"Found {total_rows:,} molecules to process.")
# --- Main Loop with TQDM ---
with tqdm(total=total_rows, desc="Total Progress", unit="mol") as pbar:
for df_test in pd.read_csv(test_file, chunksize=chunk_size):
protein_onehot = onehot_encoder.transform(df_test['protein_name'].values.reshape(-1, 1))
def feature_generator():
with Pool(processes=num_processes) as pool:
with tqdm(total=len(df_test), desc=f"Chunk Processing", leave=False, unit="mol") as inner_pbar:
ecfp_iterator = pool.imap_unordered(smiles_to_ecfp_worker, df_test['molecule_smiles'])
for i, (ecfp, protein) in enumerate(zip(ecfp_iterator, protein_onehot)):
inner_pbar.update(1)
yield ecfp + protein.tolist()
# --- CORRECTED CODE ---
# Convert the generator to a list before passing it to the model.
# This is memory-safe because it only contains data for the current chunk.
X_test_chunk = list(feature_generator())
probabilities = rf_model.predict_proba(X_test_chunk)[:, 1]
del protein_onehot
del X_test_chunk # Explicitly delete the list to free memory
gc.collect()
output_df = pd.DataFrame({'id': df_test['id'], 'binds': probabilities})
write_header = not os.path.exists(output_file)
output_df.to_csv(output_file, index=False, mode='a', header=write_header)
pbar.update(len(df_test)) # Update the main progress bar
print("Processing complete. Submission file created at 'ecfps_submission.csv'")
Using 38 worker processes. Calculating total number of molecules... Found 1,674,896 molecules to process.
Total Progress: 100%|██████████| 1674896/1674896 [19:12<00:00, 1453.05mol/s]
Processing complete. Submission file created at 'ecfps_submission.csv'
# Read the output file to verify
output_df = pd.read_csv(output_file)
print(f"Output file contains {len(output_df)} rows.")
output_df.head()
Output file contains 1674896 rows.
| id | binds | |
|---|---|---|
| 0 | 295246830 | 0.28 |
| 1 | 295246831 | 0.22 |
| 2 | 295246832 | 0.31 |
| 3 | 295246833 | 0.28 |
| 4 | 295246834 | 0.39 |