Predicting Protein Expression from CITE-seq Data
This project utilizes a wide-and-deep Multi-Layer Perceptron (MLP) built in Keras to predict surface protein expression from CITE-seq gene expression data.
The model processes input features derived from TruncatedSVD and a curated set of protein-coding genes through a series of dense layers with SELU activation and dropout. A distinctive feature is its concatenation of outputs from all intermediate layers, creating a rich feature representation that is fed into a final linear output layer, directly optimizing for the Pearson correlation metric via a custom loss function.
Future work could explore more advanced architectures like Transformers or attention mechanisms to better capture gene interdependencies, employing autoencoders for more sophisticated feature extraction, and implementing ensemble methods to enhance predictive accuracy and robustness.

Sparse Data Generation and Dimension Reduction with Truncated SVD
This notebook demonstrates how to convert 10x Chromium Single Cell Multiome ATAC raw outputs into sparse matrix representations and then apply Truncated Singular Value Decomposition (SVD) to reduce dimensionality for downstream analysis.
Multi-Omics Pattern Discovery and Classification/Regression: In the context of multi-omics pattern discovery and supervised modeling, methods like DIABLO from the MixOmics suite and MOFA have become instrumental.
DIABLO (Data Integration Analysis for Biomarker discovery using Latent cOmponents) constructs latent components that maximize covariance between different omics blocks, enabling the identification of multi-omics biomarkers for classification or regression tasks.
On the other hand, MOFA (Multi-Omics Factor Analysis) employs a Bayesian factor analysis framework to extract both shared and modality-specific factors, providing an unsupervised representation that can be readily used for downstream classification or regression by leveraging the learned factor scores as features.
Prediction of Multiome Gene Expression from ATAC-seq with High Correlation
This notebook presents a Keras-based wide-and-deep multilayer perceptron (MLP) model for predicting gene expression (RNA levels) from chromatin accessibility (ATAC-seq peaks) in the Multiome dataset from the Kaggle Open Problems - Multimodal Single-Cell Integration competition, utilizing dimension reduction via singular value decomposition (SVD) to handle sparse data—reducing inputs to 40 components and targets to 512—followed by 3-fold group k-fold cross-validation grouped by donor, achieving an out-of-fold Pearson correlation of 0.9560 and mean squared error of 10.2398, indicating strong performance in capturing the relationship between the modalities.
Future directions could include exploring advanced architectures like transformers or graph neural networks to better model genomic interactions, integrating additional single-cell modalities such as CITE-seq for multimodal fusion, fine-tuning hyperparameters with Bayesian optimization, testing on external datasets for generalizability, and incorporating biological priors like gene regulatory networks to enhance interpretability and prediction accuracy.
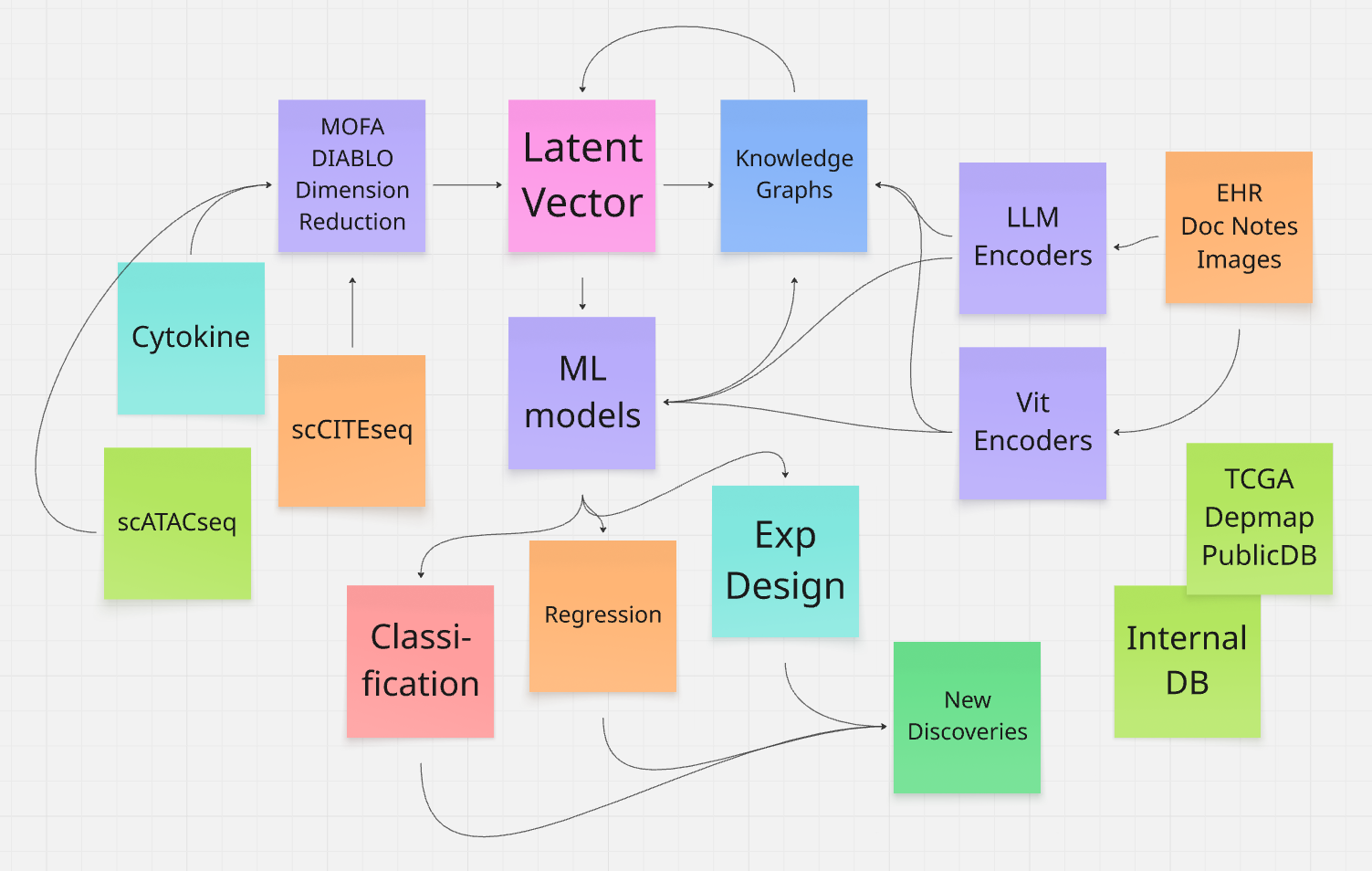
AI-Enhanced Multi-Omics Framework for Accelerating CART Therapy Development
This proposal outlines an AI-driven, patient-centric framework to integrate and analyze multi-omic data from AstraZeneca's cell therapy programs, aiming to predict outcomes, identify biomarkers, and uncover mechanistic insights in the tumor microenvironment.
Structured in four iterative phases, it begins with foundational data harmonization using tools like MOFA+ for unsupervised integration, advances to supervised predictive modeling with DIABLO and XGBoost for explainable biomarker discovery, constructs a dynamic Tumor Microenvironment Knowledge Graph (TME-KG) incorporating public databases like TCGA and DepMap for hypothesis generation, and leverages LLMs to embed unstructured clinical notes for enriched analysis.
By synthesizing high-dimensional single-cell RNA-seq, TCR-seq, cytokines, and metadata, the plan promises to de-risk clinical trials, enhance therapy persistence and safety, and foster novel discoveries, all while scaling with evolving datasets for sustained impact in oncology.
TCGA Multi-Omic Analysis of Breast Cancer Subtypes with DIABLO
This notebook offers a detailed guide to multi-omics integration via the DIABLO method in the mixOmics R package, applied to breast TCGA data including miRNA, mRNA, and proteomics to classify subtypes (Basal, Her2, LumA).
It begins with data loading and pairwise sparse PLS models for inter-omics correlation assessment, then configures a design matrix and tunes components and features through cross-validation to refine the sparse PLS-DA model. Visualizations like correlation plots, sample projections, circos diagrams, and image maps reveal feature links and subtype distinctions, while evaluations via error rates, AUC-ROC, and test predictions underscore predictive power and biomarker insights.
Drawing on breast cancer biology, it showcases DIABLO's ability to reveal multi-omics signatures for enhanced subtype classification and discovery, with reproducibility via seeded runs and session info.

Simulating Gata1 Knockout Effects on Hematopoiesis using CellOracle
Title: Simulating Gata1 Knockout Effects on Hematopoiesis using CellOracle This notebook demonstrates an in silico perturbation analysis to investigate the role of the transcription factor Gata1 in hematopoiesis. Using the CellOracle library, the notebook simulates the knockout of the Gata1 gene to predict its impact on cell fate decisions and regulatory networks. The analysis begins by loading a pre-processed single-cell RNA sequencing dataset of hematopoietic cells and the corresponding gene regulatory networks (GRNs). Subsequently, it fits predictive models to simulate the effects of Gata1 knockout, aiming to recapitulate its known functions in myeloid progenitor cell fate and erythroid differentiation.